Dall’articolo che ha pubblicato a luglio su Science Translational Medicine e dal titolo della relazione che terrà fra un mese a St Martin (Barriers to HIV Eradication), credo si possa capire quanto le questioni di “farmacomatematica” lo stiano tenendo occupato e invece – purtroppo! – quante poche soddisfazioni gli stiano arrivando sul fronte dei farmaci antilatenza (sta sperimentando il disulfiram, è vero, ma temo che ci creda poco lui per primo).
Mi pare comunque utile riprendere il post che gli avevo dedicato lo scorso luglio e, a seguire, riportare la brevissima sintesi di Lafeuillade della lezione alla University of Maryland.
************************************************
È uscito proprio ieri [13 luglio 2011] su Science Translational Medicine il frutto di almeno tre anni di lavoro di Lin Shen, all'inizio specializzando, ora collaboratore di Siliciano. Purtroppo non riesco ad avere l’articolo, ma ne è uscita una recensione su Sciencenews e ve la sintetizzo, poiché quando parla Siliciano vale sempre la pena di starlo ad ascoltare e, in questo caso, insegna a guardare a una vecchia idea con occhi nuovi (che in genere è quello che fanno gli scienziati “rivoluzionari” e che, in termini tecnici, possiamo chiamare “slittamento di significato”).
Spiegata l’efficacia variabile dei cocktail di farmaci anti-HIV
Aumentare le dosi funziona nei momenti critici del ciclo virale del virus
Rachel Ehrenberg – mercoledì 13 luglio
Un nuovo modello matematico sul funzionamento dei farmaci contro l’HIV spiega in termini biologici le variazioni – ad oggi considerate sconcertanti - nel successo delle terapie. La ricerca, pubblicata il 13 luglio su Science Translational Medicine, potrebbe aiutare ad affinare le terapie contro l’HIV e altri virus, quali quello dell’epatite C.
Il nuovo modello spiega che aumentare leggermente la dose di alcuni farmaci anti-HIV ha un grande effetto nel caso in cui questi farmaci siano pensati per colpire obiettivi molteplici. “Scoprire che più proiettili possono colpire più obiettivi può sembrare scontato – dice Robert Siliciano – ma arrivarci ha richiesto un cambiamento nel modo di pensare a una vecchissima idea: quella della relazione fra la dose di un farmaco e il suo effetto”.
Per secoli, l’efficacia di un farmaco è stata rappresentata visivamente con quella che è chiamata la “curva dose-risposta”. Questa relazione sovente, quando è rappresentata in un grafico, prende una forma a “S” allungata. Ma nel 2008 Lin Shen, che allora era uno specializzando di Siliciano, capì che la ripidità dell’inclinazione della “S” – la sua pendenza – varia in funzione delle diverse classi di farmaci anti HIV: se sale gradualmente, significa che degli aumenti nella concentrazione del farmaco migliorano la risposta in modo graduale; invece, una pendenza molto ripida significa che piccoli aumenti nella concentrazione del farmaco potrebbero distruggere molte più molecole-target.
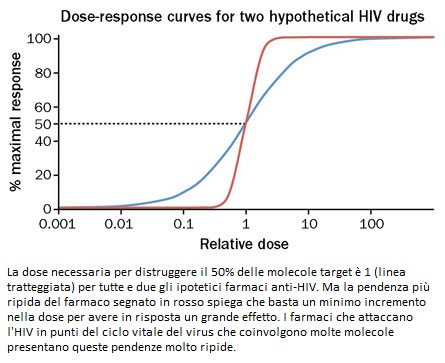
Siliciano spiega che è stupefacente il fatto che la ripidità di questa pendenza abbia qualcosa a che vedere con i farmaci contro l’HIV e che le differenze in ordine di grandezza siano davvero enormi (per esempio, aumentare il dosaggio dei più efficaci fra gli inibitori della proteasi può renderli miliardi di volte più potenti contro il virus, mentre aumentare la quantità di AZT potrebbe avere un effetto di sole 10 volte più grande rispetto a quello ottenuto con un dosaggio più basso).
Aumenti incrementali nella dose che producono un grande miglioramento nella risposta sono un fenomeno che di solito si verifica con farmaci che attaccano una molecola target in molteplici siti, e questo effetto è noto come “legame cooperativo” [dove la cooperazione dipende proprio dall’interazione che si crea fra i diversi siti]. Tuttavia, l’HIV ha un unico sito in cui il farmaco può attaccarsi, quindi dare di più di un farmaco non necessariamente lo rende più efficace.
Quel che hanno capito Siliciano e Shen, invece, è che IN CERTI MOMENTI DEL CICLO VITALE DELL’HIV, C’È COSÌ TANTO VIRUS O COSÌ TANTI APPARATI VIRALI DA ATTACCARE CHE I FARMACI CREANO UN LEGAME COOPERATIVO, MA PER COLPIRE MOLTISSIMI OBIETTIVI PIUTTOSTO CHE MOLTI SITI IN UN UNICO OBIETTIVO.
“Non era affatto scontato – dice Siliciano. Un conto è un pensiero lineare, che ti porta a vedere a quali concentrazioni di un farmaco ottieni il 50% di inibizione. Ma il virus si replica esponenzialmente: ogni cellula infetta rilascia abbastanza virus da infettare altre 10 cellule. Quindi dovevamo imparare a pensare in questi termini”.
Siliciano e Shen hanno testato il modello creando dei virus che offrissero un numero di target diverso rispetto al solito, per esempio virus che non producessero il solito numero di enzimi proteasi. Quando hanno infettato delle cellule renali con questi virus modificati e hanno calcolato le curve dose-risposta, le pendenze erano diverse rispetto a quelle dell’HIV non modificato: si è scoperto che se il virus alterato presentava al farmaco meno enzimi funzionanti da disabilitare, il virus veniva inibito a un dosaggio più basso.
Nell’abstract si precisa che questa INIBIZIONE COOPERATIVA viene spiegata da un modello in cui l’infettività richiede la partecipazione di molteplici copie del target del farmaco in un determinato stadio del ciclo vitale del virus. Coerentemente con le osservazioni sperimentali, questo modello fa delle predizioni per le diverse combinazioni di antiretrovirali, spiega la grande attività antivirale di due importanti classi di farmaci e definisce una caratteristica che rende i target dei farmaci dei “buoni” target: la cooperatività intermolecolare.
In un commento pubblicato sulla stessa rivista, Steven Deeks sostiene che i concetti esposti in questo nuovo modello aiuteranno a combattere l’HIV, ma anche altri virus, tipo l’HCV: “l’HIV è una macchina per replicare. Muta costantemente e il sistema immunitario fatica a controllarlo. Proprio perché il virus è così efficace nel fare quel che fa, non siamo mai riusciti a capire perché le combinazioni di farmaci abbiano funzionato per così tanto tempo. Ora che la matematica ci ha spiegato il segreto di questo successo dei farmaci, finalmente l’abbiamo capito”.
Fonte:
- sciencenews.org
Abstract:
- Siliciano - Lin Shen: A Critical Subset Model Provides a Conceptual Basis for the High Antiviral Activity of Major HIV Drùgs
- Perelson - Deeks: Drùg Effectiveness Explained: The Mathematics of Antiviral Agents for HIV
